HiggsToTauTau analysis: parallel
Overview
Teaching: 10 min
Exercises: 20 minQuestions
Challenge: write the HiggsToTauTau analysis parallel workflow and run it on REANA
Objectives
Develop a full HiggsToTauTau analysis workflow using parallel language
Overview
We have seen examples of full DAG-aware workflow languages (Snakemake and Yadage) and how they can be used to describe and run the RooFit example and a simple version of HiggsToTauTau example.
In this episode we shall see how to efficiently apply parallelism to speed up the HiggsToTauTau example via the scatter-gather paradigm introduced in the previous episode.
HiggsToTauTau analysis
Let us start by defining the overall skeleton of the analysis workflow.
The overall reana.yaml for this parallel analysis in Yadage looks like:
inputs:
files:
- steps.yaml
- workflow.yaml
parameters:
files:
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/GluGluToHToTauTau.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/VBF_HToTauTau.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/DYJetsToLL.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/TTbar.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W1JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W2JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W3JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/Run2012B_TauPlusX.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/Run2012C_TauPlusX.root
cross_sections:
- 19.6
- 1.55
- 3503.7
- 225.2
- 6381.2
- 2039.8
- 612.5
- 1.0
- 1.0
short_hands:
- [ggH]
- [qqH]
- [ZLL,ZTT]
- [TT]
- [W1J]
- [W2J]
- [W3J]
- [dataRunB]
- [dataRunC]
workflow:
type: yadage
file: workflow.yaml
outputs:
files:
- fit/fit.png
Note that the input files, cross-sections, and short names are defined as arrays. These are the arrays we will scatter over.
The overall reana.yaml for this parallel analysis in Snakemake looks like:
inputs:
files:
- Snakefile
workflow:
type: snakemake
file: Snakefile
outputs:
files:
- fit/fit.png
Note that this reana.yaml file is very minimal: we are basically only declaring that we shall use
Snakemake workflow type, and all the details will live in the Snakefile that will be defining the
workflow. The Snakefile will start by defining parameters:
uri = "root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced"
files = [
"GluGluToHToTauTau",
"VBF_HToTauTau",
"DYJetsToLL",
"DYJetsToLL",
"TTbar",
"W1JetsToLNu",
"W2JetsToLNu",
"W3JetsToLNu",
"Run2012B_TauPlusX",
"Run2012C_TauPlusX",
]
cross_sections = [
19.6,
1.55,
3503.7,
3503.7,
225.2,
6381.2,
2039.8,
612.5,
1.0,
1.0,
]
short_hands = [
"ggH",
"qqH",
"ZLL",
"ZTT",
"TT",
"W1J",
"W2J",
"W3J",
"dataRunB",
"dataRunC",
]
Note how we have defined files, cross_sections, and short_hands representing various data
sets that we shall be processing in parallel. We shall use these arrays as Snakemake wildcards
over which the computations will be distributed in a parallel manner. The short_hands array
gives each dataset a compact label used in downstream rules and output filenames.
Note also that DYJetsToLL appears twice on purpose: the same input dataset is processed under
two different selections, ZLL (Z → ℓℓ) and ZTT (Z → ττ). Because Snakemake aligns the three
arrays element-by-element, the file and its cross-section are repeated so that each selection
gets its own scatter slot.
We can also declare the desired overall final outputs of the workflow:
rule all:
input:
"fit/fit.png",
"plot/pt_met.png"
The rest of the Snakefile will be discussed below.
HiggsToTauTau skimming
The skimming step definition looks like:
- name: skim
dependencies: [init]
scheduler:
scheduler_type: multistep-stage
parameters:
input_file: {step: init, output: files}
cross_section: {step: init, output: cross_sections}
output_file: '{workdir}/skimmed.root'
scatter:
method: zip
parameters: [input_file, cross_section]
step: {$ref: 'steps.yaml#/skim'}
where the step is defined as:
skim:
process:
process_type: 'interpolated-script-cmd'
script: |
./skim {input_file} {output_file} {cross_section} 11467.0 0.1
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
skimmed_file: '{output_file}'
Note the scatter paradigm that will cause nine parallel jobs for each input dataset file.
rule skim:
output:
"skim/{files}_{cross_sections}.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p skim && cd /analysis/skim && ./skim {uri}/{wildcards.files}.root $workspace/{output} {wildcards.cross_sections} 11467.0 0.1"
HiggsToTauTau histogramming
The histograms can be produced as follows:
- name: histogram
dependencies: [skim]
scheduler:
scheduler_type: multistep-stage
parameters:
input_file: {stages: skim, output: skimmed_file}
output_names: {step: init, output: short_hands}
output_dir: '{workdir}'
scatter:
method: zip
parameters: [input_file, output_names]
step: {$ref: 'steps.yaml#/histogram'}
with:
histogram:
process:
process_type: 'interpolated-script-cmd'
script: |
for x in {output_names}; do
python histograms.py {input_file} $x {output_dir}/$x.root;
done
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
glob: true
publish:
histogram_file: '{output_dir}/*.root'
rule histogram:
input:
"skim/{files}_{cross_sections}.root"
output:
"histogram/{files}_{cross_sections}_{short_hands}.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p histogram && cd /analysis/skim && python histograms.py $workspace/{input} {wildcards.short_hands} $workspace/{output}"
HiggsToTauTau merging
Time to gather! How do we merge scattered results?
- name: merge
dependencies: [histogram]
scheduler:
scheduler_type: singlestep-stage
parameters:
input_files: {stages: histogram, output: histogram_file, flatten: true}
output_file: '{workdir}/merged.root'
step: {$ref: 'steps.yaml#/merge'}
with:
merge:
process:
process_type: 'interpolated-script-cmd'
script: |
hadd {output_file} {input_files}
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
merged_file: '{output_file}'
rule merge:
input:
expand("histogram/{files}_{cross_sections}_{short_hands}.root", zip, files=files, cross_sections=cross_sections, short_hands=short_hands)
output:
"merge/merged.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"mkdir -p merge && hadd {output} {input}"
HiggsToTauTau fitting
The fit can be performed as follows:
- name: fit
dependencies: [merge]
scheduler:
scheduler_type: singlestep-stage
parameters:
histogram_file: {step: merge, output: merged_file}
fit_outputs: '{workdir}'
step: {$ref: 'steps.yaml#/fit'}
with:
fit:
process:
process_type: 'interpolated-script-cmd'
script: |
python fit.py {histogram_file} {fit_outputs}
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-statistics-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
fit_results: '{fit_outputs}/fit.png'
rule fit:
input:
"merge/merged.root"
output:
"fit/fit.png"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-statistics-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p fit && cd /fit && python fit.py $workspace/{input} $workspace/fit"
HiggsToTauTau plotting
Challenge time! Add plotting step to the workflow.
Exercise
Following the example above, write plotting step and plug it into the overall workflow.
Solution
The addition to the workflow specification is:
- name: plot
dependencies: [merge]
scheduler:
scheduler_type: singlestep-stage
parameters:
histogram_file: {step: merge, output: merged_file}
plot_outputs: '{workdir}'
step: {$ref: 'steps.yaml#/plot'}
The step is being defined as:
plot:
process:
process_type: 'interpolated-script-cmd'
script: |
python plot.py {histogram_file} {plot_outputs} 0.1
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
fitting_plot: '{plot_outputs}'
rule plot:
input:
"merge/merged.root"
output:
"plot/pt_met.png"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p plot && cd /analysis/skim && python plot.py $workspace/{input} $workspace/plot 0.1"
Full workflow
We are now ready to assemble the previous stages together and run the example on the REANA cloud.
Exercise
Write and run the HiggsToTauTau parallel workflow on REANA cloud. How many jobs does the workflow have? How much faster is it executed compared to the simple serial version?
Solution
The REANA specification file reana.yaml looks as follows:
inputs:
files:
- steps.yaml
- workflow.yaml
parameters:
files:
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/GluGluToHToTauTau.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/VBF_HToTauTau.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/DYJetsToLL.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/TTbar.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W1JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W2JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/W3JetsToLNu.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/Run2012B_TauPlusX.root
- root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced/Run2012C_TauPlusX.root
cross_sections:
- 19.6
- 1.55
- 3503.7
- 225.2
- 6381.2
- 2039.8
- 612.5
- 1.0
- 1.0
short_hands:
- [ggH]
- [qqH]
- [ZLL, ZTT]
- [TT]
- [W1J]
- [W2J]
- [W3J]
- [dataRunB]
- [dataRunC]
workflow:
type: yadage
file: workflow.yaml
outputs:
files:
- fit/fit.png
The workflow definition file workflow.yaml is:
stages:
- name: skim
dependencies: [init]
scheduler:
scheduler_type: multistep-stage
parameters:
input_file: {step: init, output: files}
cross_section: {step: init, output: cross_sections}
output_file: '{workdir}/skimmed.root'
scatter:
method: zip
parameters: [input_file, cross_section]
step: {$ref: 'steps.yaml#/skim'}
- name: histogram
dependencies: [skim]
scheduler:
scheduler_type: multistep-stage
parameters:
input_file: {stages: skim, output: skimmed_file}
output_names: {step: init, output: short_hands}
output_dir: '{workdir}'
scatter:
method: zip
parameters: [input_file, output_names]
step: {$ref: 'steps.yaml#/histogram'}
- name: merge
dependencies: [histogram]
scheduler:
scheduler_type: singlestep-stage
parameters:
input_files: {stages: histogram, output: histogram_file, flatten: true}
output_file: '{workdir}/merged.root'
step: {$ref: 'steps.yaml#/merge'}
- name: fit
dependencies: [merge]
scheduler:
scheduler_type: singlestep-stage
parameters:
histogram_file: {step: merge, output: merged_file}
fit_outputs: '{workdir}'
step: {$ref: 'steps.yaml#/fit'}
- name: plot
dependencies: [merge]
scheduler:
scheduler_type: singlestep-stage
parameters:
histogram_file: {step: merge, output: merged_file}
plot_outputs: '{workdir}'
step: {$ref: 'steps.yaml#/plot'}
The workflow steps defined in steps.yaml are:
skim:
process:
process_type: 'interpolated-script-cmd'
script: |
./skim {input_file} {output_file} {cross_section} 11467.0 0.1
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
skimmed_file: '{output_file}'
histogram:
process:
process_type: 'interpolated-script-cmd'
script: |
for x in {output_names}; do
python histograms.py {input_file} $x {output_dir}/$x.root;
done
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
glob: true
publish:
histogram_file: '{output_dir}/*.root'
merge:
process:
process_type: 'interpolated-script-cmd'
script: |
hadd {output_file} {input_files}
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
merged_file: '{output_file}'
fit:
process:
process_type: 'interpolated-script-cmd'
script: |
python fit.py {histogram_file} {fit_outputs}
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-statistics-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
fit_results: '{fit_outputs}/fit.png'
plot:
process:
process_type: 'interpolated-script-cmd'
script: |
python plot.py {histogram_file} {plot_outputs} 0.1
environment:
environment_type: 'docker-encapsulated'
image: gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3
imagetag: master
publisher:
publisher_type: interpolated-pub
publish:
fitting_plot: '{plot_outputs}'
The REANA specification file reana.yaml looks as follows:
inputs:
files:
- Snakefile
workflow:
type: snakemake
file: Snakefile
outputs:
files:
- fit/fit.png
The workflow definition file Snakefile is:
uri = "root://eospublic.cern.ch//eos/root-eos/HiggsTauTauReduced"
files = [
"GluGluToHToTauTau",
"VBF_HToTauTau",
"DYJetsToLL",
"DYJetsToLL",
"TTbar",
"W1JetsToLNu",
"W2JetsToLNu",
"W3JetsToLNu",
"Run2012B_TauPlusX",
"Run2012C_TauPlusX",
]
cross_sections = [
19.6,
1.55,
3503.7,
3503.7,
225.2,
6381.2,
2039.8,
612.5,
1.0,
1.0,
]
short_hands = [
"ggH",
"qqH",
"ZLL",
"ZTT",
"TT",
"W1J",
"W2J",
"W3J",
"dataRunB",
"dataRunC",
]
rule all:
input:
"fit/fit.png",
"plot/pt_met.png"
rule skim:
output:
"skim/{files}_{cross_sections}.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p skim && cd /analysis/skim && ./skim {uri}/{wildcards.files}.root $workspace/{output} {wildcards.cross_sections} 11467.0 0.1"
rule histogram:
input:
"skim/{files}_{cross_sections}.root"
output:
"histogram/{files}_{cross_sections}_{short_hands}.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p histogram && cd /analysis/skim && python histograms.py $workspace/{input} {wildcards.short_hands} $workspace/{output}"
rule merge:
input:
expand("histogram/{files}_{cross_sections}_{short_hands}.root", zip, files=files, cross_sections=cross_sections, short_hands=short_hands)
output:
"merge/merged.root"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"mkdir -p merge && hadd {output} {input}"
rule fit:
input:
"merge/merged.root"
output:
"fit/fit.png"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-statistics-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p fit && cd /fit && python fit.py $workspace/{input} $workspace/fit"
rule plot:
input:
"merge/merged.root"
output:
"plot/pt_met.png"
container:
"docker://gitlab-registry.cern.ch/awesome-workshop/awesome-analysis-eventselection-stage3:master"
shell:
"workspace=$(pwd) && mkdir -p plot && cd /analysis/skim && python plot.py $workspace/{input} $workspace/plot 0.1"
Results
The computational graph of the workflow looks like:
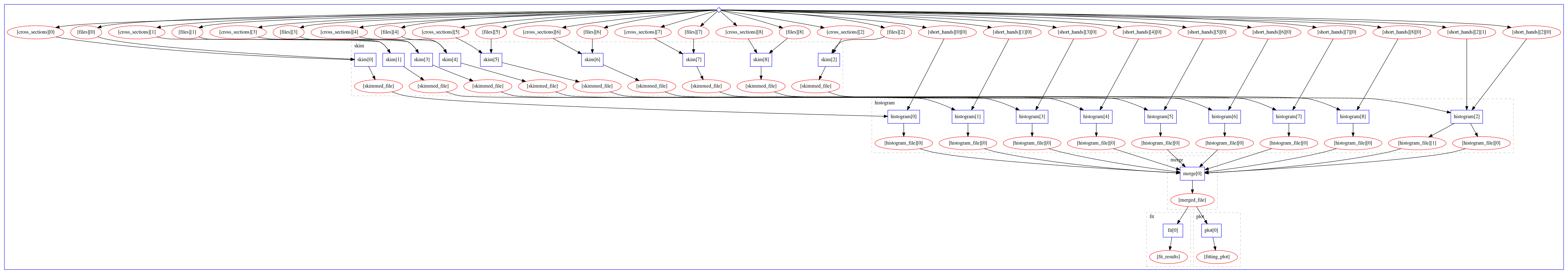
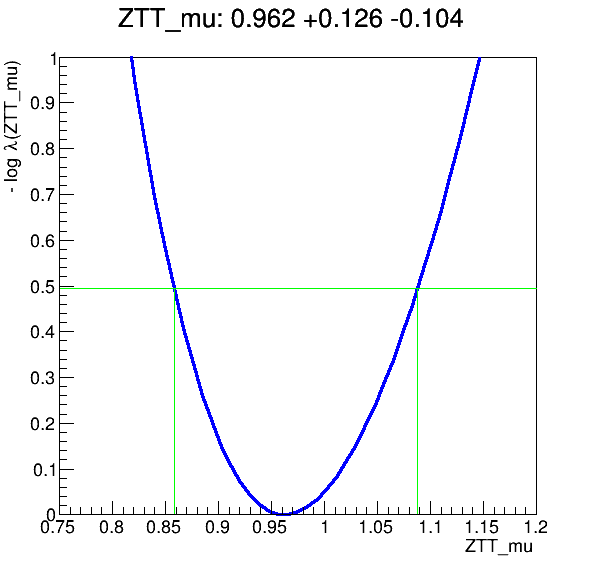
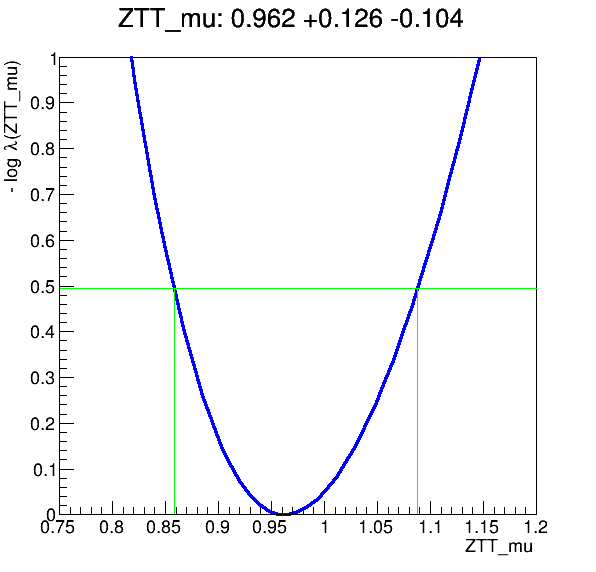
The workflow produces the following fit:
The dependency graph of the workflow rules, defined in Snakefile, looks like:
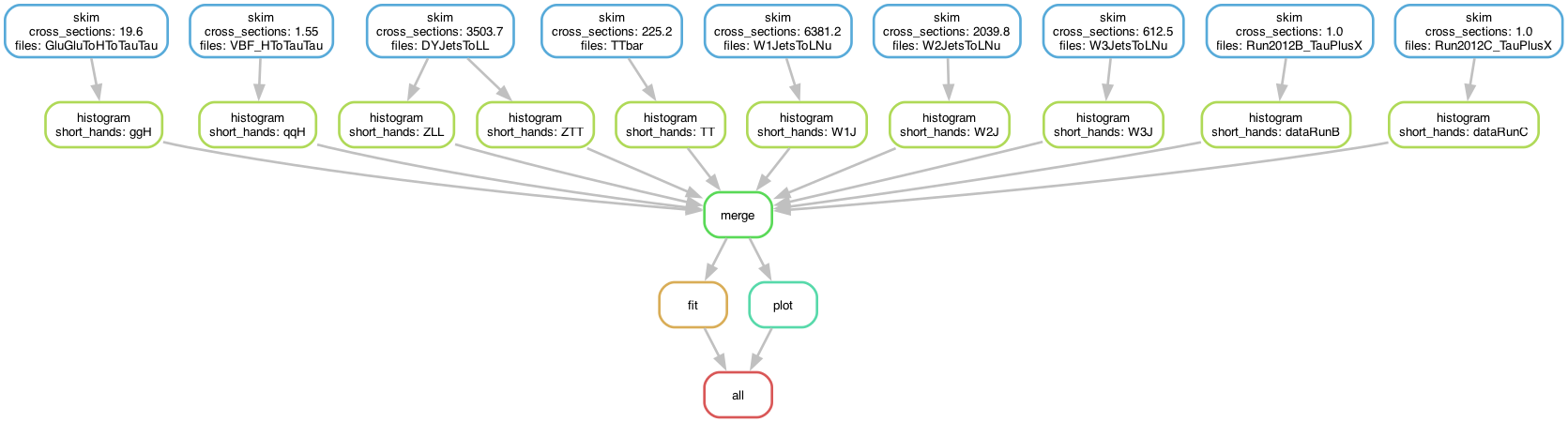
The skimming and histogramming calculations were parallelised over various input files, leading to the following runtime computational graph:
The workflow produces the following fit:
Key Points
Use step dependencies to express main analysis stages
Use scatter-gather paradigm in stages to massively parallelise DAG workflow execution
REANA usage scenarios remain the same regardless of workflow language details