Analysis automation with Snakemake
Learning Objectives
Learn what analysis automation is and how it helps with analysis preservation
Learn how to create a pipeline with Snakemake
Documentation and environments
You can find full documentation for Snakemake here; you can also ask any questions you have on the ~reproducible channel on mattermost. Web searches and Stack Overflow are also your friends.
LHCb specific: using lb-conda environment
Snakemake is best-run at LHCb using the lb-conda environment.
This environment comes with very recent versions of
ROOT, python, cmake, g++, snakemake, etc., ready to use.
In general, it is recommended that if you are running non-lhcb software
(e.g., code you’ve written yourself for your analysis)
it should be done with lb-conda.
More information on using lb-conda can be found here.
To have access to lb-conda you must first have sourced LbEnv.
This is done by default on lxplus;
otherwise, it is done with source /cvmfs/lhcb.cern.ch/lib/LbEnv
(assuming cvmfs is installed).
The default environment can be entered with the command lb-conda default,
where default is the name of the environment.
This will enter you into an interactive bash shell.
The full list of available environments can be found by running
lb-conda --list.
You can optionally pass a command after the environment name,
e.g., lb-conda default foo,
which will run the foo command in the environment and then exit.
(This is similar to the behaviour of lb-run.)
If you want, you can include your .bashrc file in the created environment
by running lb-conda default bash -c 'bash --rcfile ~/.bashrc'.
Be careful if you do this–it can lead to conflicts in the environment.
You can check if Snakemake is working by calling snakemake --help
in the lb-conda default environment.
Workflow preservation
There is an ongoing effort at LHCb to ensure analysis workflows are preserved. You can learn more about this effort and how Snakemake figures into it here.
Basic Tutorial
What is a workflow?
When performing an analysis, you typically start with some set of input files containing the data you are interested in and want to end up with some set of output files containing the results of your measurements. A workflow is what gets you from the input to the output.
Let us consider a simple example. Create a new directory to work in and generate some fake input files:
mkdir basic_tutorial
cd basic_tutorial
mkdir input
touch input/{a..z}.in
ls input/
You should see a list of empty files named a.in, b.in, …, and z.in.
Perform the workflow
Add the text My name is a. to the end of input/a.in
and save it as output/a.out without modifying input/a.in.
Do this for each file in input/.
Solution
You could open each file, type the appropriate text
(e.g., My name is a., My name is b., etc.),
and save it as the appropriate output file,
but since this is such a simple workflow,
we can easily make the shell do it for us:
mkdir -p output # make the directory if needed
for file in input/*.in; do
noext="${file%.in}" # drop the extension
name="${noext##input/}" # drop the directory
ofile="output/${name}.out" # declare output file name
cat "$file" > "$ofile" # create output file with contents of input file
echo "My name is $name." >> "$ofile" # append the required text
done
Congratulations, you’ve just defined and executed a workflow!
Why use a workflow management system?
“The Snakemake workflow management system is a tool to create reproducible and scalable data analyses”
The example workflow above is very simple, but you can probably imagine more complicated ones. Suppose you wanted to adjust your input files in more complicated ways, had multiple types of input or output files, or wanted to do calculations that didn’t depend on just one input file. Or suppose that your calculations took a long time and you only wanted to re-run them if the inputs had changed. These are all reasons to use a workflow management system.
A workflow management system allows you to:
Keep a record of how your scripts are used and what their input dependencies are
Run multiple steps in sequence, parallelising where possible
Automatically detect if something changes and then reprocess data if needed
Using a workflow management system forces you to:
Keep your code and your locations in order
Structure your code so that it is user-independent
Standardise your scripts
Bonus: Standardised scripts can sometimes be used across analyses!
If you have ever performed an HEP analysis or looked at the code for someone else’s analysis, you probably understand why the above features are so useful.
Introducing Snakemake
Snakemake allows you to create a set of rules,
each one defining a step of your analysis.
The rules need to be written in a file called Snakefile.
For each rule you need to provide:
The input: Data files, scripts, executables or any other files.
The expected output. It’s not required to list all possible outputs. Just those that you want to monitor or that are used by a subsequent step as inputs.
A command to run to process the input and create the output.
The basic rule is:
rule myname:
input: "myinput1", "myinput2"
output: "myoutput"
shell: "Some command to go from in to out"
Let’s give it a try with our simple example above.
In the same basic_tutorial directory,
let us first remove all our hard work by running
rm -rf output/
Now, we create our Snakefile.
In your favorite text editor,
open a new file called Snakefile and type
rule name_files:
input: "input/a.in"
output: "output/a.out"
shell: "cat input/a.in > output/a.out && echo 'My name is a.' >> output/a.out"
Snakemake now knows how to create output/a.out.
You can run it by calling
snakemake output/a.out --cores 1
This tells Snakemake that you want to generate output/a.out if needed
and to use only one computing core to do so.
When you run it,
you should see something like:
Building DAG of jobs...
Using shell: /cvmfs/lhcbdev.cern.ch/conda/envs/default/2021-09-07_04-06/linux-64/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count min threads max threads
---------- ------- ------------- -------------
name_files 1 1 1
total 1 1 1
Select jobs to execute...
[Mon Nov 15 18:30:25 2021]
rule name_files:
input: input/a.in
output: output/a.out
jobid: 0
resources: tmpdir=/tmp/username
[Mon Nov 15 18:30:26 2021]
Finished job 0.
1 of 1 steps (100%) done
Complete log: /afs/cern.ch/user/u/username/basic_tutorial/.snakemake/log/2021-11-15T183022.449488.snakemake.log
Here, we see that Snakemake selected the rule name_files to create output/a.out.
Calling cat output/a.out,
we see that the expected file was created with the appropriate text.
Notice that we did not have to tell Snakemake to create the output/ directory;
Snakemake took care of it automatically.
Now, run snakemake output/a.out --cores 1 again.
You should see output like:
Building DAG of jobs...
Nothing to be done.
Complete log: /afs/cern.ch/user/u/username/basic_tutorial/.snakemake/log/2021-11-15T183344.711303.snakemake.log
Since output/a.out already exists, Snakemake did not try to create it again.
We still have files b-z to create.
We could create 25 more rules, one for each of the remaining files,
but Snakemake provides a feature called wildcards so we don’t have to.
We edit our Snakefile to read
rule name_files:
input: "input/{name}.in"
output: "output/{name}.out"
shell: "cat {input} > {output} && echo 'My name is {wildcards.name}.' >> {output}"
Take a moment to understand what is happening in this rule declaration.
Wildcards are defined in output
and can then be accessed in input and shell.
All wildcards must be present in output.
We are essentially telling Snakemake that this is a rule that can create
files of the form output/{name}.out;
we cannot specify wildcards in input that are not present in output
because Snakemake will not know what they should be.
Wildcard syntax
Notice that the wildcard name appears as {name}
in the declaration of input and output
but as {wildcards.name} in the declaration of shell.
This is an unavoidable quirk of how Snakemake works,
and you must remember it to avoid errors.
Snakemake can now handle all of our input and output. Run
snakemake output/{a..z}.out --cores 4
and notice that Snakemake has created all the output, just as we did manually before.
Snakemake has many features that our original solution, simply running a shell script, does not, but one is particularly obvious in this case: Since we told Snakemake to run with 4 cores, it created (up to) 4 output files at once. We didn’t have to wait for each output file to be created one-by-one; we were able to take advantage of our computing power to perform multiple tasks at the same time. This built-in parallelism is one of the nicest features of using a workflow manager.
How many cores should I use?
You can see how many cores are on the machine you’re using by calling
nproc from the command line.
If you’re on lxplus, there should be 10.
If you want Snakemake to use all of them,
you can use --cores all.
This is bad practice on a shared machine, like on lxplus;
you should leave some computing resources available to other users
so that they can at least log in.
We can tell Snakemake which files to create from within our Snakefile. At the beginning of the file, add
rule name_all:
input: [f"output/{chr(x)}.out" for x in range(ord('a'), ord('z') + 1)]
We have declared a new rule, name_all, at the beginning of the file
with all our required output files listed as input
and no output files declared.
Snakemake interprets the first rule in the Snakefile as the default rule,
and Snakemake will run it if no other output is requested.
If we remove output/,
rm -r output/
and run Snakemake again, this time without specifying any output,
snakemake --cores 4
we see that output/ is produced the same as before.
Snakemake and python
Notice that we have used python list-comprehension
to declare the input files for rule name_all.
Snakemake is python-based,
so you can execute arbitrary python code anywhere in your Snakefile
if you want to.
Types of input
Notice that we have given a list as the input to rule name_all
instead of a file name.
Snakemake can interpret many types of input,
including strings, tuples, lists, and even functions!
See the documentation here.
Congratulations, you’ve just defined a workflow using Snakemake!
Re-running rules
We saw above that if an output file already exists,
Snakemake will not create it again.
If you call snakemake --cores 4 now,
you should see that it exits without running anything,
since all the files in output/ already exist.
Now, open input/a.in and add some text:
I have been modified.
Run Snakemake again.
Now, you should see that Snakemake runs name_files just once to create output/a.out.
It does not try to create any of the other files,
and it creates output/a.out even though it already exists.
This is because we listed input/a.in as an input;
since it had been modified since the creation of output/a.out,
Snakemake decided output/a.out needed to be updated
and ran name_files again.
We can force the re-running of rules
using the --force or -f command-line argument.
Calling
snakemake output/a.out --cores 4 -f
will tell Snakemake to create output/a.out regardless of modification times.
If we want to re-run the whole workflow, we can use --forceall.
Always do a dry run!
Since Snakemake relies on modification times to decide which rules to run, using version control software (such as git) can become complicated, as files are routinely modified and then returned to their original form. You can find a helpful solution for this problem here.
Regardless of whether you use version control,
always do a dry run before executing your workflow!
You can do this by calling Snakemake with
the --dry-run or -n command-line argument,
which will print the rules Snakemake would execute
but prevents them from actually being executed.
If you have many rules and don’t want to fill up your terminal with output,
you can use the --quiet or -q command-line argument in addition to -n
to print just a summary.
It’s worth reiterating because this can easily become an issue
for complicated workflows
and it will save you a lot of wasted time:
Always run with -n first!
Chaining rules
We can also tell our Snakefile how to create our input files.
In our simple example here,
our inputs are just empty files in the input/ directory,
which we created above by calling touch input/{a..z}.in.
Let’s create a rule to do this for us.
Create the input
Add a rule to Snakefile that can create
input/a.in, input/b.in, …, and input/z.in.
Solution
Add a rule like the following to your Snakefile:
rule create_input:
output: "input/{name}.in"
shell: "touch {output}"
You can name it anything you like and use any wildcard you want; it will work regardless.
To check your solution, run
rm -rf input/ output/
snakemake --cores 4
Snakemake should create both the input and the output.
Take a moment to understand what has happened.
We told Snakemake we wanted to generate output/a.out, …, output/z.out,
and it knew to create input/a.in, …, input/z.in
because the rule name_files depends on them.
We can make this dependency more explicit.
Modify name_files to read
rule name_files:
input: rules.create_input.output
output: "output/{name}.out"
shell: "cat {input} > {output} && echo 'My name is {wildcards.name}.' >> {output}"
As long as it is declared after rule create_input
and all of the wildcards of create_input are also declared in name_files,
this should produce the same output as before.
Notice that we could also just ask Snakemake to produce input/a.in
without reqesting output/a.out;
as far as Snakemake is concerned,
input/a.in is just another file it knows how to produce.
The limits of wildcards
Our Snakefile
isn’t limited to creating output/a.out, …, output/z.out.
Trying calling
snakemake output/hello_world.out --cores 4
and observe what happens.
Snakemake has created input/hello_world.in and output/hello_world.out.
Now try
snakemake output/foo.bar --cores 4
This time, Snakemake raises a MissingRuleException;
this is because the extension .bar doesn’t match the output
for rule name_files, which expects the .out extension.
Allow arbitrary output file extensions
Teach Snakefile how to create an output file with any file extension.
Solution
Modify name_files to read:
rule name_files:
input: rules.create_input.output
output: "output/{name}.{ext}"
shell: "cat {input} > {output} && echo 'My name is {wildcards.name}.' >> {output}"
Notice that this doesn’t affect the input file extension,
which is still .in.
Now, if you run
snakemake output/foo.bar --cores 4
Snakemake should generate input/foo.in and output/foo.bar
As you can see from this challenge,
Snakemake allows input and output files to be of any type;
you could be creating .root files, .gif files, or .docx files
and Snakemake will execute regardless.
You can see the whole working Snakefile here.
Key Points
Explain what a workflow is.
Explain some of the benefits of using a workflow management system.
Define a workflow using Snakemake.
Advanced Tutorial
In this tutorial, we will examine more advanced Snakemake topics using pre-defined inputs.
Prerequisites
To begin, run
mkdir advanced_tutorial
cd advanced_tutorial
wget https://github.com/hsf-training/analysis-essentials/raw/master/snakemake/code/advanced_tutorial/input.tar
tar -xvf input.tar
ls input/
You should see address.txt and phone.txt.
The first contains names followed by addresses;
the second contains names followed by phone numbers.
These are the pre-defined input files
You can find the ultimate solution (how your workflow might look after solving all of the challenges in this tutorial) here.
Get Luca’s address and phone number
Write a Snakefile with a single rule that creates an output file
with Luca’s address and phone number.
Hint: the command grep lists all lines in a file containing a given text.
Solution
Your Snakefile could look something like this:
rule Luca_info:
input: "input/address.txt", "input/phone.txt"
output: "output/Luca/info.txt"
shell: "grep Luca {input[0]} > {output} && grep Luca {input[1]} >> {output}"
Notice that we specified multiple input files and referred to them by index. We could also have named them:
rule Luca_info:
input:
address="input/address.txt",
phone="input/phone.txt",
output: "output/Luca/info.txt"
shell: "grep Luca {input.address} > {output} && grep Luca {input.phone} >> {output}"
Get Luca’s address and phone number in three steps
Remember that one of the nice things about using Snakemake is that we can run things in parallel instead of sequentially. Modify your Snakefile to get Luca’s address and phone number in separate rules, then merge them into a single output file.
Solution
Your Snakefile could look something like this:
rule merge_data:
input:
address="output/Luca/address.txt",
phone="output/Luca/phone.txt",
output: "output/Luca/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_address:
input: "input/address.txt"
output: "output/Luca/address.txt"
shell: "grep Luca {input} > {output}"
rule get_phone:
input: "input/phone.txt"
output: "output/Luca/phone.txt"
shell: "grep Luca {input} > {output}"
This is not much better than our original solution,
since we still end up running two sequential bash commands in the final step,
but one can easily imagine this being helpful if the rules
get_address and get_phone took a long time.
In the solution above,
we don’t really need the output of get_address and get_phone;
they are just waypoints on our way to the output of merge_data
and we could delete them when we finished.
Snakemake provides a way to do this for us, using the temp utility function:
rule merge_data:
input:
address="output/Luca/address.txt",
phone="output/Luca/phone.txt",
output: "output/Luca/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_address:
input: "input/address.txt"
output: temp("output/Luca/address.txt")
shell: "grep Luca {input} > {output}"
rule get_phone:
input: "input/phone.txt"
output: temp("output/Luca/phone.txt")
shell: "grep Luca {input} > {output}"
This tells Snakemake to remove the specified output once all the rules
that use it as an input are finished.
Note that you can only mark output as temporary in this way, not input.
Use wildcards
Modify your Snakefile to get anyone’s address and phone number. Tell it to get Luca’s, Fred’s, and Guillaume’s by default.
Solution
Your Snakefile could look something like this:
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_address:
input: "input/address.txt"
output: temp("output/{name}/address.txt")
shell: "grep {wildcards.name} {input} > {output}"
rule get_phone:
input: "input/phone.txt"
output: temp("output/{name}/phone.txt")
shell: "grep {wildcards.name} {input} > {output}"
The Snakemake workflow can be represented by a directed acyclic graph (DAG). In the above solution for just Luca, it looks something like this:
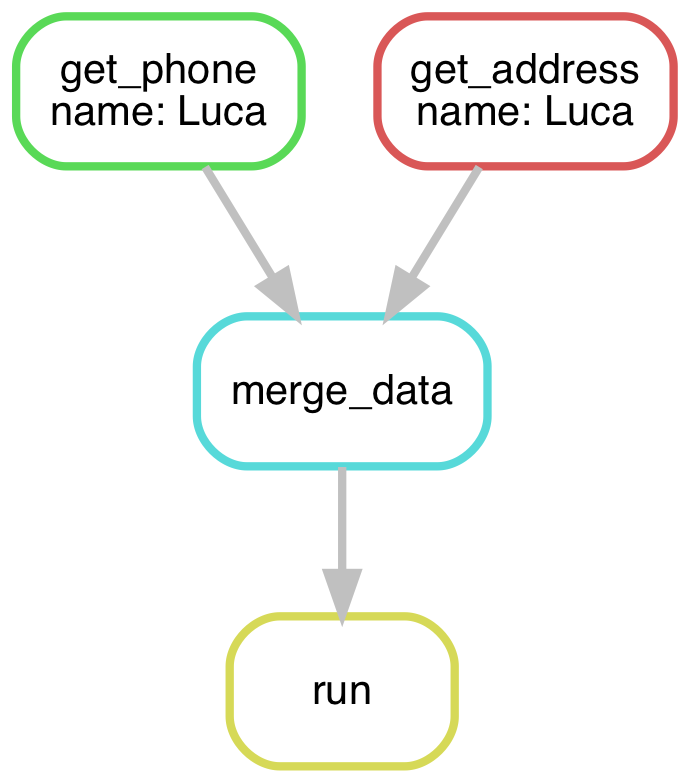
In the most recent solution for Luca, Fred, and Guillaume, it looks more like this:
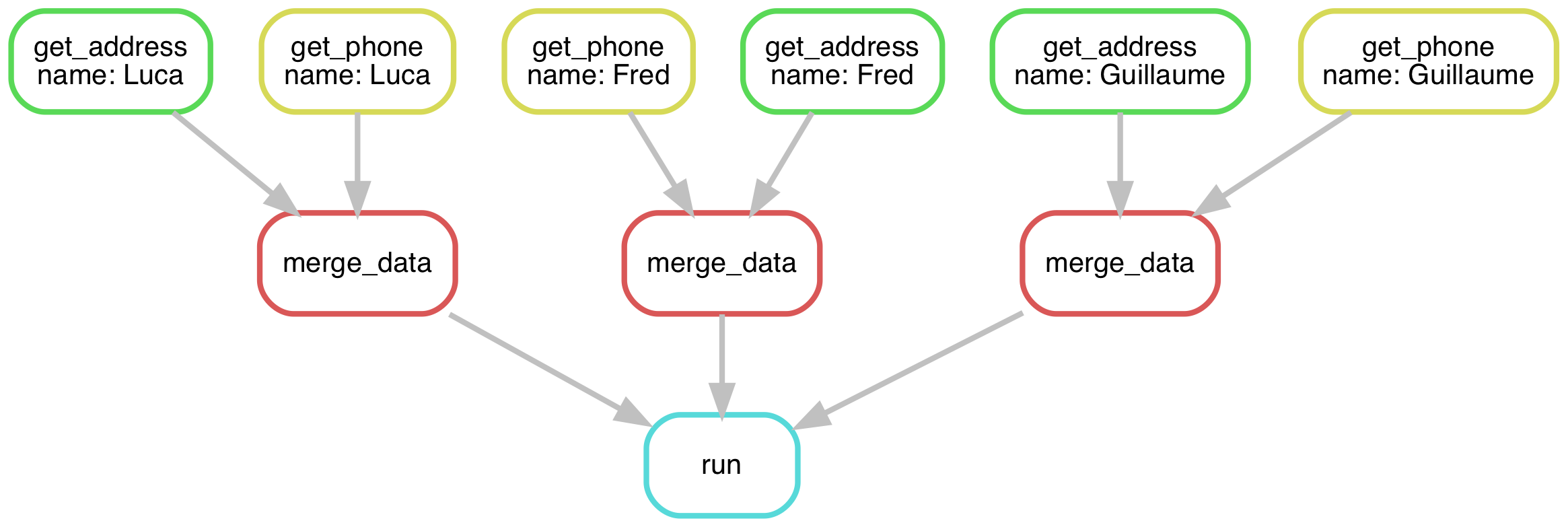
Use more wildcards
Reduce your Snakefile to contain just three rules.
Solution
Your Snakefile could look something like this:
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_info:
input: "input/{info}.txt"
output: temp("output/{name}/{info}.txt")
shell: "grep {wildcards.name} {input} > {output}"
Since the rules get_address and get_phone were so nearly identical,
we simply combined them into a single rule called get_info using wildcards.
Running scripts
Snakemake can execute anything in the command-line, including calling python scripts.
Call python scripts
Modify your workflow to look up anyone’s address and phone number and write them in a single output file without including their name.
Solution
Instead of using shell commands,
we can write a python file, get_info.py:
"""Extract information about a person."""
import argparse
parser = argparse.ArgumentParser()
parser.add_argument("name", help="person of interest")
parser.add_argument("infile", help="text file with information")
parser.add_argument("outfile", help="where to write extracted information")
args = parser.parse_args()
# -- find information
found = None
with open(args.infile) as f:
for ln in f:
if ln.startswith(args.name):
found = ln
break # assume only one line with info
info = found[len(args.name):]
# -- write information
with open(args.outfile, "w") as f:
f.write(info)
Your Snakefile could then look something like this:
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_info:
input:
exe="get_info.py",
infile="input/{info}.txt",
output: temp("output/{name}/{info}.txt")
shell: "python {input.exe} {wildcards.name} {input.infile} {output}"
Notice that we included get_info.py as an input.
If you modify it (add an additional comment, for example),
Snakemake will run the rule get_info again.
You do not have to list it as an input if you do not want this behaviour;
in such a case, you would drop it from the input declarations
and modify the shell command to read
python get_info.py {wildcards.name} {input.infile} {output}
Listing dependencies
Your rule can depend on files that you don’t list as input;
Snakemake just won’t know about them.
How many of the dependencies to list as input
is a matter of personal preference.
You will certainly want to include any input data files.
In the example above,
the python script is included as a dependency,
which means that Snakemake will run rule get_info if it changes.
This is probably the behaviour that you want–after all,
if you change how to create the output file,
it should be recreated.
The tricky part is if you find yourself making a lot of minor changes.
Suppose you update the documentation in a file
without changing any of the actual commands;
Snakemake will still re-run the rule–it has no way of knowing
whether the changes you made were substantial or inconsequential.
Also, notice that you could make substantial changes to the shell command
in a rule of your Snakefile and Snakemake would not re-run it
(unless you listed your Snakefile as an input,
which is not recommended).
One philosophy is to avoid listing the executable code as an input
and to remember to re-run the changed rules manually
(most easily done using the --forcerun or -R
argument).
This has the benefit of treating changes to rule definitions
and changes to executable files equally,
that is, having Snakemake ignore them both.
The downside, of course, is that if you have a complicated DAG,
this is not always easy to keep track of,
and besides, one of the reasons to use Snakemake is
so you don’t have to remember such things.
In practice, the solution is subjective and situation-dependent.
You probably don’t want to list your numpy distribution as an input,
you probably do want to list your python executable as one.
To avoid re-running rules just because you changed a comment,
you could use touch -m to adjust the modification time of the file;
you could creatively use --allowed-rules
to avoid specific rules;
or, if you are using git,
you could use a combination of --date
and the solution here.
These workarounds all require you to be careful, however,
and can lead to unexpected results.
The cleanest solution is to just live with the fact that you will have to re-run rules after minor changes.
You have two ways to specify commands.
One is shell, which we’ve been using.
The other is run that instead directly takes python code.
For example,
the previous solution could be written
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_info:
input:
infile="input/{info}.txt",
output: temp("output/{name}/{info}.txt")
run:
found = None
with open(input.infile) as f:
for ln in f:
if ln.startswith(wildcards.name):
found = ln
break # assume only one line with info
info = found[len(wildcards.name):] # assume space follows name
with open(str(output), "w") as f:
f.write(info)
f.write("\n")
You can also mix and match by calling
shell
from within the python code.
Instead of shell or run,
you can also declare
script,
which is similar to run but it allows you to refer to an external file.
Log files
Within Snakemake, a log file is a special type of output file that is not deleted if a rule fails.
Snakemake and execution failures
If an error is raised during the execution of a rule,
Snakemake deletes the files listed as output,
since they could be corrupted.
To see this happen,
simply add the line assert False to the end of get_info.py
In practice, log files allow you to monitor the behaviour of your rules and debug when things go wrong. To declare a log file, simply add it to your rule declaration:
rule myname:
input: "myinput1", "myinput2"
log: "mylog"
output: "myoutput"
shell: "Some command to go from in to out and create log"
Just as with output,
you must supply a command that produces a log file.
The shell redirect command &> is particularly useful here;
it captures both stdout and stderr and saves them in a (recreated) file.
You can read more about redirection in the bash manual
here.
Note that this redirection is only available if you are declaring
shellactions in your rule declarations; if you are usingrunorscript, you might consider using theloggingpackage.
Add log files
Modify your workflow to print progress updates and save them in log files.
Solution
First, we modify get_info.py to print status updates:
"""Extract information about a person."""
import argparse
parser = argparse.ArgumentParser()
parser.add_argument("name", help="person of interest")
parser.add_argument("infile", help="text file with information")
parser.add_argument("outfile", help="where to write extracted information")
args = parser.parse_args()
# -- find information
print("extracting information...")
found = None
with open(args.infile) as f:
for ln in f:
if ln.startswith(args.name):
found = ln
break # assume only one line with info
if not found:
raise ValueError(f"{args.infile} contains no info on '{args.name}'")
info = found[len(args.name):]
print("found information")
# -- write information
print("writing information...")
with open(args.outfile, "w") as f:
f.write(info)
print("done")
Your Snakefile could then look something like this:
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_info:
input:
exe="get_info.py",
infile="input/{info}.txt",
log: "log/{name}/{info}.txt"
output: temp("output/{name}/{info}.txt")
shell: "python {input.exe} {wildcards.name} {input.infile} {output} &> {log}"
Notice that log includes all the wildcards used in output;
this ensures that every instance of the rule has a unique log file.
If you run Snakemake with --forceall,
you should see your log files created in the log/ directory.
Config files
Often you want to run the same rule on different samples or with different options for your scripts. This can be done in snakemake using config files written in yaml.
For example let’s declare some data files in a cfg.yaml file:
data:
- 'data1.root'
- 'data2.root'
Now in your Snakefile you can load this config file and then its content will be available to the rules as a dictionary called “config”. Yes, it seems black magic, but it works! Your Snakefile will look something like this:
configfile: '/path/to/cfg.yaml'
rule dosomething:
input:
exe='mycode.py',
data=config['data'],
output: ['plot1.pdf', 'plot2.pdf']
shell: "python {input.exe} {input.data} {output}"
The config dictionary can be used anywhere, also inside the shell command or even outside a rule.
Make a config file
Prerequisites
Download some alternate input files:
wget https://github.com/hsf-training/analysis-essentials/raw/master/snakemake/code/advanced_tutorial/input_alt.tar
tar -xvf input_alt.tar
ls input_alt/
You should see a new set of addresses and phone numbers.
Create a config file that can specify whether to use
the addresses and phone numbers in input/ or those in input_alt/.
Solution
First, we create cfg.yaml:
data: input_alt
Then, modify your Snakefile to look something like this:
configfile: "cfg.yaml"
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
rule get_info:
input:
exe="get_info.py",
infile=f"{config['data']}/{{info}}.txt",
log: "log/{name}/{info}.txt"
output: temp("output/{name}/{info}.txt")
shell: "python {input.exe} {wildcards.name} {input.infile} {output} &> {log}"
Notice that we have escaped the braces around
infoin the declaration ofinput.infile; this is because we have used an f-string, which uses braces to determine which items to replace. Putting double braces aroundinfotells the interpreter that we want the braces to remain after the f-string substitution is complete, allowing Snakemake to identifyinfoas a wildcard.
Be careful–unless you explicitly declare your config file as an input,
Snakemake will not automatically rerun a rule when it changes.
You will have to remember to force it to run.
Includes
The Snakefile can quickly grow to a monster with tens of rules.
For this reason it’s possible to split them into more files
and then include them into the Snakefile.
For example you might have a fit_rules.snake and efficiency_rules.snake
and then your Snakefile will look like this:
include: "/path/to/fit_rules.snake"
include: "/path/to/efficiency_rules.snake"
Use includes
Move your rules to other files and include them.
Solution
You could create merge.snake,
rule merge_data:
input:
address="output/{name}/address.txt",
phone="output/{name}/phone.txt",
output: "output/{name}/info.txt"
shell: "cat {input.address} > {output} && cat {input.phone} >> {output}"
and get.snake,
rule get_info:
input:
exe="get_info.py",
infile=f"{config['data']}/{{info}}.txt",
log: "log/{name}/{info}.txt"
output: temp("output/{name}/{info}.txt")
shell: "python {input.exe} {wildcards.name} {input.infile} {output} &> {log}"
Your modified Snakefile would then look like this:
configfile: "cfg.yaml"
include: "merge.snake"
include: "get.snake"
rule run:
input: ["output/Luca/info.txt", "output/Fred/info.txt", "output/Guillaume/info.txt"]
Key Points
Explain a DAG and how
Snakemakerules can relate to each other.Create a
Snakefilethat can run existing scripts.Keep track of progress and problems using log files.
Explain the benefits of
includein Snakemake and how to use them.